Hereditary cystic kidney diseases represent one of the most important causes of end stage renal disease in childhood. The main representatives are autosomal recessive polycystic kidney disease (ARPKD), nephronophthisis and related ciliopathies (NPH/NPH-RC), Bardet – Biedl Syndrome and hepatocyte nuclear factor-1beta nephropathy (HNF1B). As rare diseases their incidences range from 1:5000 to 1:100000. In Germany approximately 300-450 children are affected. Most of those diseases follow an autosomal – recessive inheritance except HNF1B nephropathy which inherited in an autosomal – dominant manner.
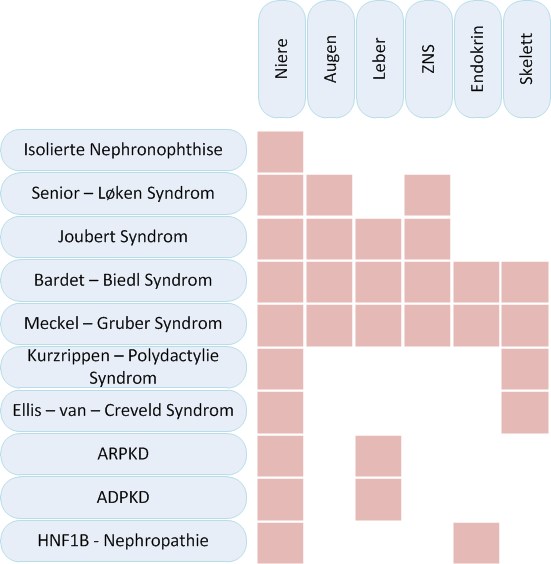
Cystic kidney diseases can affect exclusively the kidneys or they can be part of syndromic diseases that affect different other organs, especially the eyes, liver, endocrine organs or the skeleton.
During the past years far over 70 disease – causing genes could be identified. Most of those genes are responsible for the correct structure and function of primary cilia. Primary cilia are antenna-like multi-functional structures on the cell surface. They are important for cell-division, cell-orientation, fotoreception, mechanoreception and signal transduction e.g. in growth and development. (s. “of cilia and cysts“)
Despite the tremendous scientific progress in the last years, the underlying pathomechanisms are still not fully understood.
Genetic and phenotypic overlap
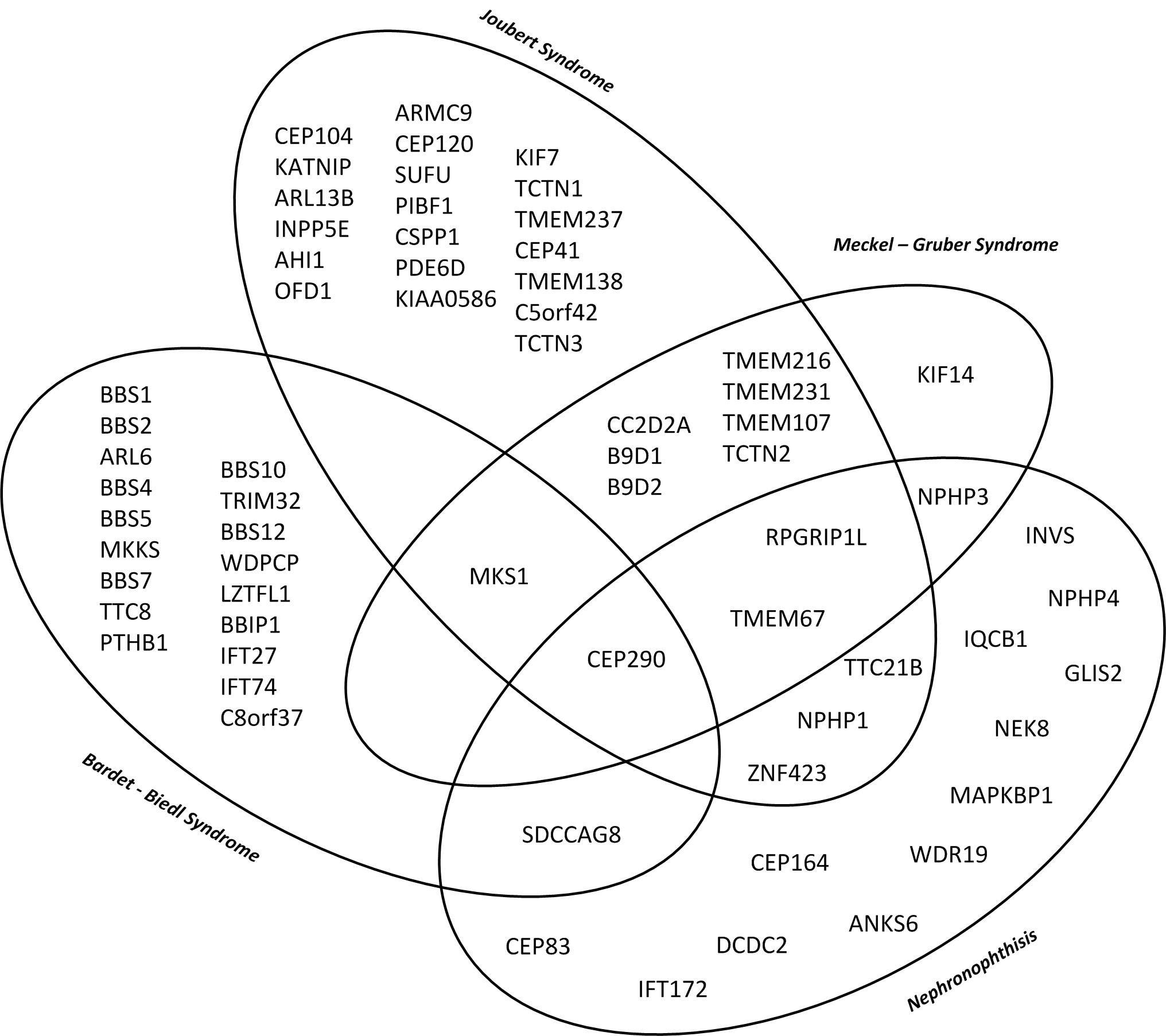
There is significant phenotypic and genotypic overlap as well as an important genotypic heterogenicity. One disease can be caused by multiple genes while at the same time dysfunction of one gene can cause different phenotypes. A variety of organs can be affected to different extents. Thus end stage renal disease can progress differently in patients with the same genetic defect.
Making the diagnosis is challanging for treating doctors as well as for patients.
This diagramme illustrates the genetic and phenotypic overlap in hereditary ciliopathies featuring cystic kidney disease from isolated nephronophthisis to lethal Meckel–Gruber Syndrome. (10)
 |
 |
Genes in early onset cystic kidney diseases
| Nephronophthisis (1,2)
(~63% rate of genetic proof) |
more than 1 % of cases | Genes accounting for less than 1 % of cases |
|
|
|
| Joubert Syndrome (3)
(62 – 94% rate of genetic proof) |
Genes accounting for more than 1 % of cases | Genes accounting for less than 1 % of cases |
|
|
|
| Bardet – Biedl Syndrome (4)
(70-80% rate of genetic proof) |
Most common genes | Less common genes |
|
|
|
| Meckel – Gruber Syndrome (5,6)
(60% rate of genetic proof) |
Most common genes | Less common genes |
|
|
|
| Senior – Løken Syndrome |
|
|
| Short rib thoracic dysplasia with or without polydactyly – including Mainzer –Saldino Syndrome (MZSDS) and Jeune asphyxiating thoracic dysplasia (ATD) (7) |
|
|
| Ellis – van – Creveld Syndrome |
|
|
| ARPKD (8) |
|
|
| ADPKD (9) |
|
|
| HNF1B – nephropathy |
|
|
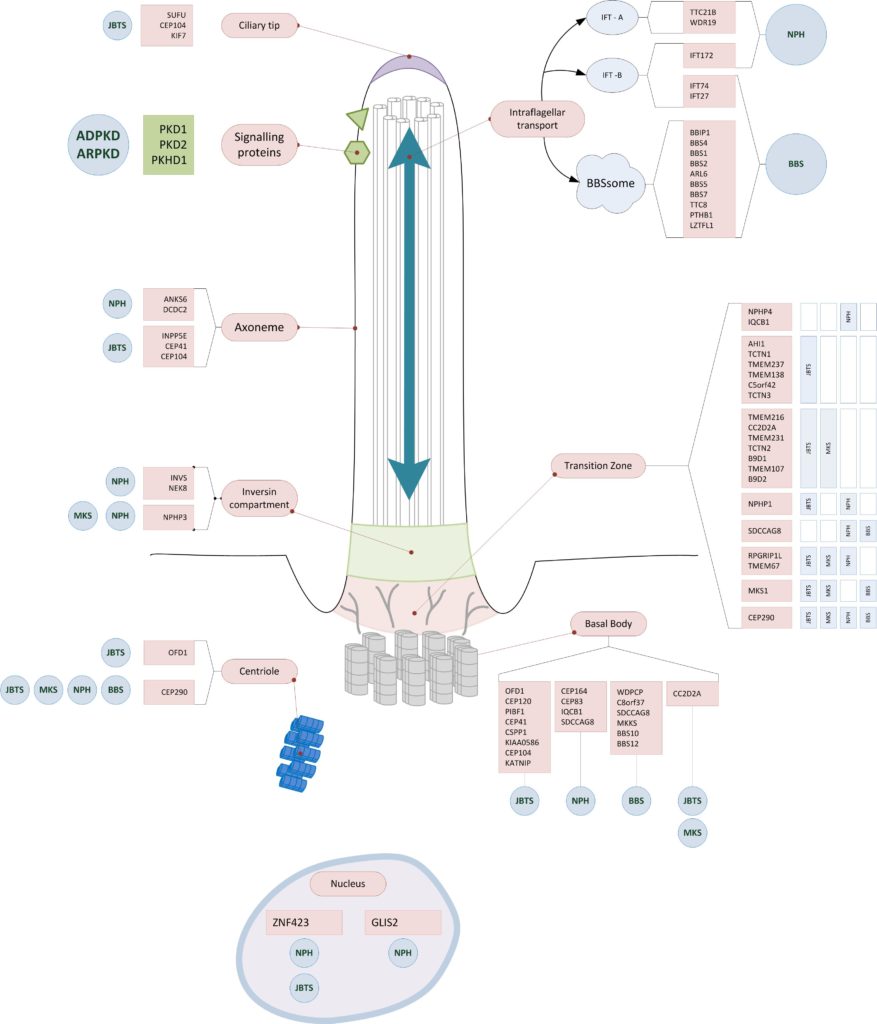
Affected structures of the primary cilium
Most of the affected genes in hereditary cystic kidney diseases encode for proteins of the primary cilium. They often have functions in the tranzition zone, the basal body or the intraflagellar transport. (10)
Sources (1) Stokman M. et. al GeneReviews® [Internet]. http://www.ncbi.nlm.nih.gov/books/NBK368475/ PMID: 27336129. (2) König J. et. al, Phenotypic Spectrum of Children with Nephronophthisis and Related Ciliopathies. Clin J Am Soc Nephrol. 2017 Dec 7;12(12):1974-1983. PMID: 29146700; (3) Parisi M. et. al, GeneReviews® [Internet]. http://www.ncbi.nlm.nih.gov/books/NBK1325/ PMID: 20301500. (4) Ece Solmaz A. et. al, Targeted multi-gene panel testing for the diagnosis of Bardet Biedl syndrome: Identification of nine novel mutations across BBS1, BBS2, BBS4, BBS7, BBS9, BBS10 genes. Eur J Med Genet. 2015 Dec;58(12):689-94. PMID: 26518167. (5) Hartill V. et. al, Meckel-Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances. Front Pediatr. 2017 Nov 20;5:244. PMID: 29209597; (6) Szymanska K. et al, Founder mutations and genotype-phenotype correlations in Meckel-Gruber syndrome and associated ciliopathies. Cilia. 2012 Oct 1;1(1):18. PMID: 23351400; (7) Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol. 2017 Mar 1;9(3). pii: a028191. PMID: 27793968; (8) Lu H. et. al, Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet. 2017 Jul;49(7):1025-1034. PMID: 28530676; (9) Cornec-Le Gall E. et. al, Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J Am Soc Nephrol. 2018 Jan;29(1):13-23. PMID: 29038287; (10) König JC. et. al, Network for Early Onset Cystic Kidney Diseases – A Comprehensive Multidisciplinary Approach to Hereditary Cystic Kidney Diseases in Childhood. Front Pediatr. 2018 Feb 13;6:24. PMID: 29497606; (11) www.omim.org