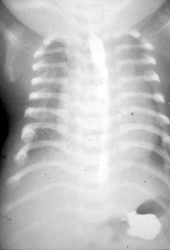
Die Jeune asphyxierende Thoraxdystrophie gehört zu der Gruppe der Kurzrippen-Polydaktylie-Syndrome (short-rib thoracic dysplasia – SRTD). Sie ist gekennzeichnet durch kurze Rippen, einen dysplastischen Thorax mit ausgeprägter Hypoplasie der Lungen sowie anderen Skelettanomalien (Skoliose, kurze Extremitäten). Zusätzliche Krankheitsmerkmale können eine Nephronophthise, fibrozystische Veränderungen an Leber und Pankreas, eine Retinopathie sowie eine Hyperbilirubinämie sein. Das Krankheitsbild variiert je nach zugrunde liegender Mutation stark, so dass einige Fälle bereits perinatal letal verlaufen können, während sich in anderen Fällen lediglich eine geringgradige Ausprägung und Beeinträchtigung zeigt. Im längeren Verlauf stellen rezidivierende pulmonale Infektionen sowie eine progrediente Niereninsuffizienz die Hauptprobleme der Erkrankung dar.
Jeune asphyxierende Thoraxdystrophie - tabellarische Zusammenfassung
| Synonyme |
|
| Hauptauffälligkeiten |
|
| Ergänzende Befunde |
|
| Manifestation | ab Geburt; oft bereits präpartal diagnostizierbar aufgrund der Thoraxhypoplasie |
| Häufigkeit | 1 : 200,000–1.000.000 Lebendgeborene |
| Vererbung | autosomal rezessiv |
| Pathogenese | zugrundeliegende Mutationen führen zu Störung des intraflagellären Transports in Zilien. |
| Verlauf, Prognose | Die neonatale Prognose ist abhängig von der Ausprägung der Lungenhypoplasie und Ateminsuffizienz. Die Letalität in den ersten Lebenswochen beträgt ca. 20-60%.
Im Verlauf bestimmt die progrediente Niereninsuffizienz die Prognose. Bei Entwicklung einer Retinitis pigmentosa ist ein progredienter Visusverlust bis zur vollständigen Erblindung zu befürchten. Die kognitive Entwicklung der Kinder ist meist unbeeinträchtigt. |
| Therapie | Keine kausale Therapie möglich. Notwendigkeit einer multidisziplinären Betreuung von Chirurgen, Pulmonologen, Nephrologen und Ophtalmologen. |
Bislang bekannte Jeune Syndrom Gene
| Krankheit | OMIM Phenotypnummer | Gen | OMIM – Gennummer | alternativer Phänotyp und OMIM Phenotypnummer | ||
| 1 | Kurzrippen-Polydaktylie Syndrom 1 | 611263 | IFT80 | 611177 | ||
| 2 | Kurzrippen-Polydaktylie Syndrom 3 | 613091 | DYNC2H1 | 603297 | ||
| 3 | Kurzrippen-Polydaktylie Syndrom 4 | 613819 | TTC21B/ IFT139 | 612014 | ||
| 4 | Kurzrippen-Polydaktylie Syndrom 5 | 614376 | WDR19/IFT144 | 608151 | NPHP13 | 614377 |
| SLSN8 | 616307 | |||||
| 5 | Kurzrippen-Polydaktylie Syndrom 6 | 263520 | NEK1 | 604588 | ||
| 6 | Kurzrippen-Polydaktylie Syndrom 7 | 614091 | WDR35 | 613602 | ||
| 7 | Kurzrippen-Polydaktylie Syndrom 8 | 615503 | WDR60 | 615462 | ||
| 8 | Kurzrippen-Polydaktylie Syndrom 9 | 266920 | IFT140 | 614620 | Retinitis pigmentosa 80 | 617781 |
| 9 | Kurzrippen-Polydaktylie Syndrom 10 | 615630 | IFT172 | 607386 | Retinitis pigmentosa 71 | 616394 |
| 10 | Kurzrippen-Polydaktylie Syndrom 11 | 615633 | WDR34 | 613363 | ||
| 11 | Kurzrippen-Polydaktylie Syndrom 13 | 616300 | CEP120 | 613446 | JBTS31 | 617761 |
| 12 | Kurzrippen-Polydaktylie Syndrom14 | 616546 | KIAA0586 | 610178 | JBTS23 | 616490 |
(01/2018)
Quellen
- Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet C Semin Med Genet. 2012;160C:165–74.
- Wolf M. Nephronophthisis and related syndromes. Curr Opin Pediatr. 2015 Apr; 27(2): 201–211.